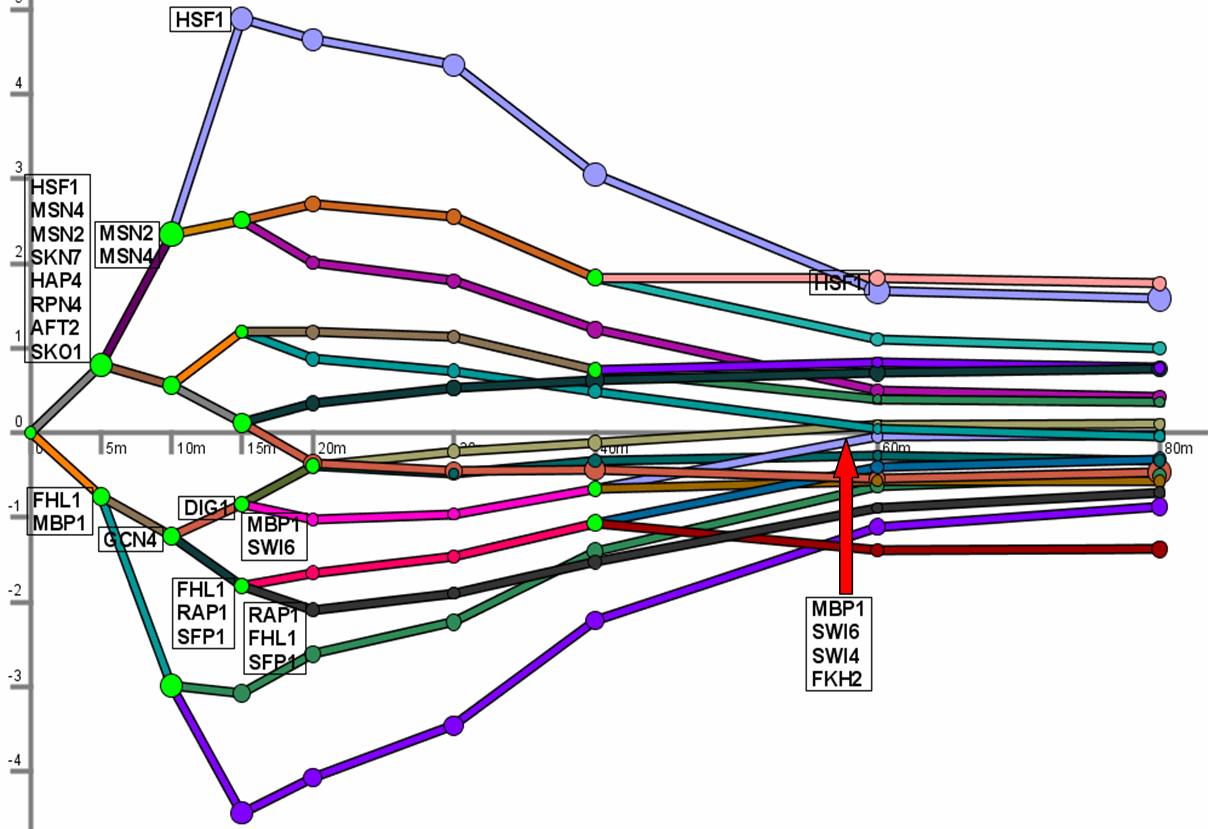
The Dynamic Regulatory Events Miner (DREM) allows one to model,
analyze, and visualize transcriptional gene regulation dynamics. The method of DREM takes as input time series gene expression data and
static transcription factor-gene interaction data (e.g. ChIP-chip data), and produces as output a dynamic regulatory map.
The dynamic regulatory map highlights major bifurcation events in the time series expression data and transcription factors potentially
responsible for them. DREM currently supports both yeast and E. coli.
See the manual and papers for more details.
The Short Time-series Expression Miner (STEM) is a Java program for clustering, comparing, and visualizing short time
series gene expression data from microarray experiments (~8 time points or fewer). STEM allows researchers to identify significant temporal
expression profiles and the genes associated with these profiles and to compare the behavior of these genes across multiple conditions. STEM
is fully integrated with the Gene Ontology (GO) database supporting GO category gene enrichment analyses for sets of genes having the
same temporal expression pattern. STEM also supports the ability to easily determine and visualize the behavior of genes belonging to
a given GO category or user defined gene set, identifying which temporal expression profiles were enriched for these genes.
(Note: While STEM is designed primarily to analyze data from short time course experiments it can be used to analyze data from any
small set of experiments which can naturally be ordered sequentially including dose response experiments.)
ContRep and DiffExp for continuous representation and differentially expressed genes in time series expression data
The ContRep and DiffExp programs are matlab implemations of methods
for continuous represenation of time series expriession datasets and for identifying differentially expressed genes in time
series experiments. The programs use mixed effcets models which utlizes co-=expressed genes to imporve the accuracy of the
reconstrcuted profiles. Differential exoression is determined based on this contrinuous representation overcoming problems related
to sampling rates and alignment. See our papers
Continuous Representations of Time Series Gene Expression Data and
Comparing the continuous representation of time-series expression profiles to identify differentially expressed genes for more details.
A Matlab implementation of ContRep with a README file can be downloaded from here.
A Matlab implementation of DiffExp with a README file can be downloaded from here.
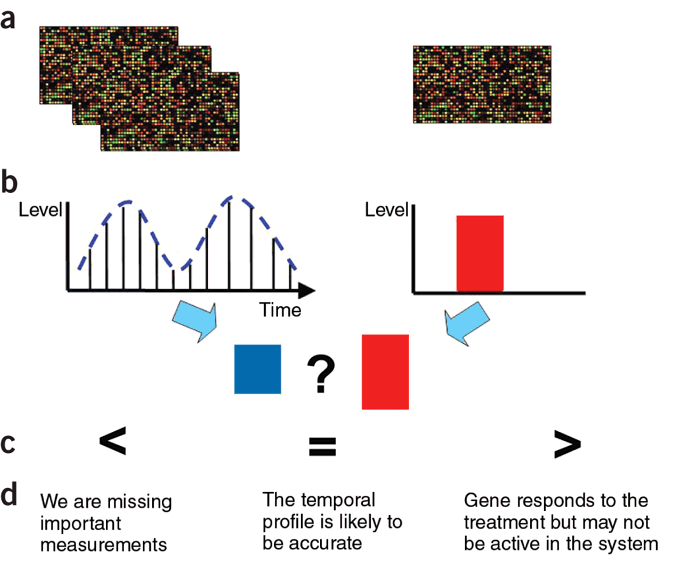
Checksum, implemented in Matlab, is a software tool for determining
if the sampling rates used for an expression experiment are appropriate. Checksum works by combining time-series and static (average)
expression data. For each gene it determines whether its temporal expression profile can be reconciled with its static expression levels.
Optimal leaf ordering (OLO)
Optima leaf ordering is a software for solving the traveling salemans problem for binary trees. The main goal of this
program is to imporve the interprability of hierarchical clustering results. Hierarchical clustering is one of the most popular methods
for clustering gene expression data. Hierarchical clustering
assembles input elements into a single tree, and subtrees
represent different clusters. Thus, using hierarchical
clustering one can analyze and visualize relationships
in scales that range from large groups (clusters) to single
genes. However, hierarchcial cluistering does not specificy how to order the two nodes that are combined at each stage. OLO finds the
optimal ordering for a given tree, that is an ordering the maximzes the sum of similarities of neighboring leafs in the ordering.
The code is based on our ISMB (Bioinformaitcs) 2001 paper and on our
Bioinformatics 2003 paper. You can download a
C++ source code implementing the
Optimal Leaf Ordering algorithm.
This program is written for the Linux operating system and its input and output are compatible with Eisen's
Cluster and TreeView software. Alternatively,
David Venet wrote a set of Matlab functions for analyzing gene expression
data. His
MatArray toolbox contains (among other useful things) an
implementation of our Optimal Leaf Ordering algorithm.